Evaluation of computational techniques for predicting non-synonymous single nucleotide variants pathogenicity
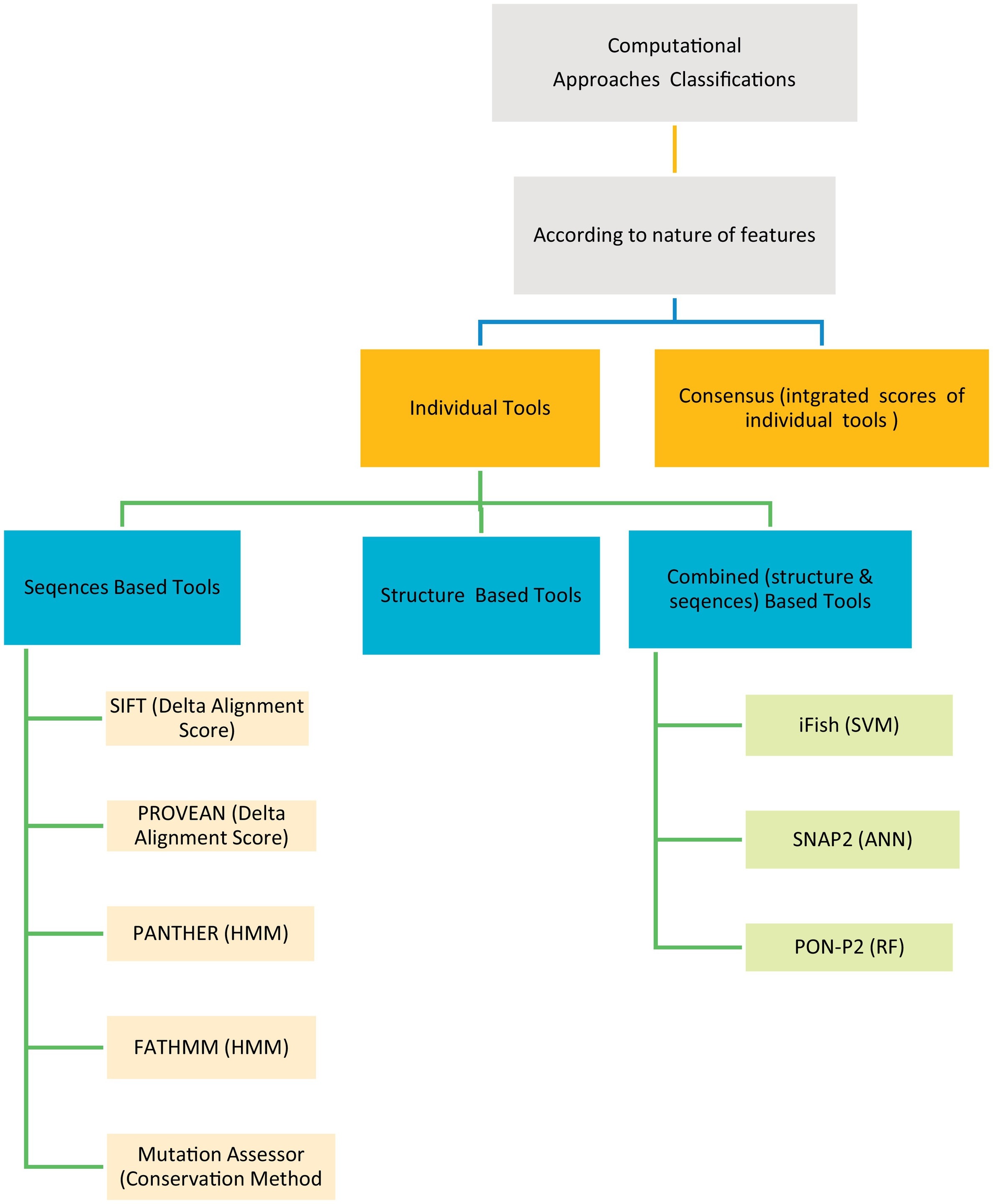
The human genetic diseases associated with many factors, one of these factors is the non-synonymous Single Nucleotide Variants (nsSNVs) cause single amino acid change with another resulting in protein function change leading to disease. Many computational techniques have been released to expect the impacts of amino acid alteration on protein function and classify mutations as pathogenic or neutral. Here in this article, we assessed the performance of eight techniques; FATHMM, SIFT, Provean, iFish, Mutation Assessor, PANTHER, SNAP2, and PON- P2 using a VaribenchSelectedPure dataset of 2144 pathogenic variants and 3777 neutral variants extracted from the free standard database “Varibench.” The first five techniques achieve (45.60–83.75) % specificity, (52.64–94.13) % sensitivity, (51.00–88.90) % AUC, and (49.76–88.24) % ACC on whole dataset, while all eight techniques achieve (36.54–77.88) % specificity, (50.00–75.00) % sensitivity, (51.00–76.40) % AUC, and (25.00–77.78) % ACC on random sample dataset. We also created a Meta classifier (CSTJ48) that combines FATHMM, iFish, and Mutation Assessor. It registers 96.33% specificity, 86.07% sensitivity, 91.20% AUC, and 91.89 ACC. By comparing the results, it's clear that FATHMM gives the highest performance over the seven individual techniques, where it achieves 83.75% and 77.88% specificity, 94.13%, and 75.00% sensitivity, 88.90% and 76.40% AUC, and 88.24% and 77.78% ACC on whole and random sample dataset, respectively. Also, the launched Meta classifier (CSTJ48) is outperforming over all the eight individual tools that compared here. © 2018 Elsevier Inc.
Related Publications

Hands-on analysis of using large language models for the auto evaluation of programming assignments