Ab initio density functional theory investigation of the interaction between carbon nanotubes and water molecules during water desalination Process
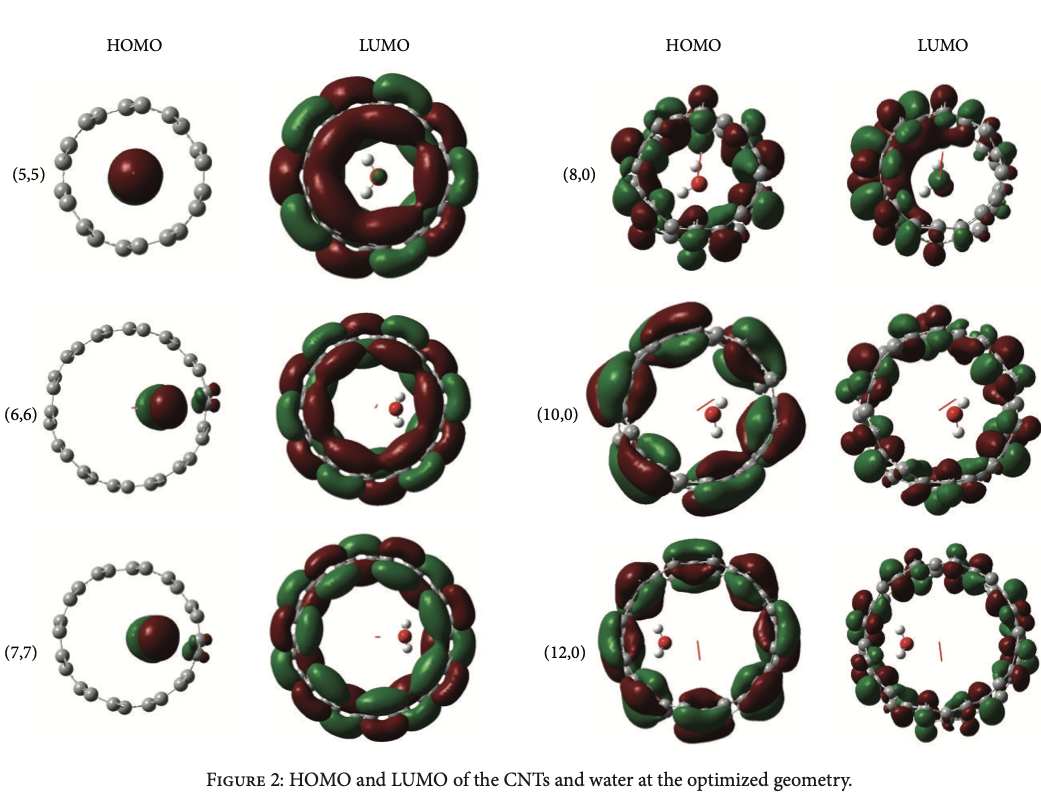
Density functional theory calculations using B3LYP/3-21G level of theory have been implemented on 6 carbon nanotubes (CNTs) structures (3 zigzag and 3 armchair CNTs) to study the energetics of the reverse osmosis during water desalination process. Calculations of the band gap, interaction energy, highest occupied molecular orbital, lowest unoccupied molecular orbital, electronegativity, hardness, and pressure of the system are discussed. The calculations showed that the water molecule that exists inside the CNT is about 2-3 Å away from its wall. The calculations have proven that the zigzag CNTs are more efficient for reverse osmosis water desalination process than armchair CNTs as the reverse osmosis process requires pressure of approximately 200 MPa for armchair CNTs, which is consistent with the values used in molecular dynamics simulations, while that needed when using zigzag CNTs was in the order of 60 MPa. © 2013 Loay A. Elalfy et al.