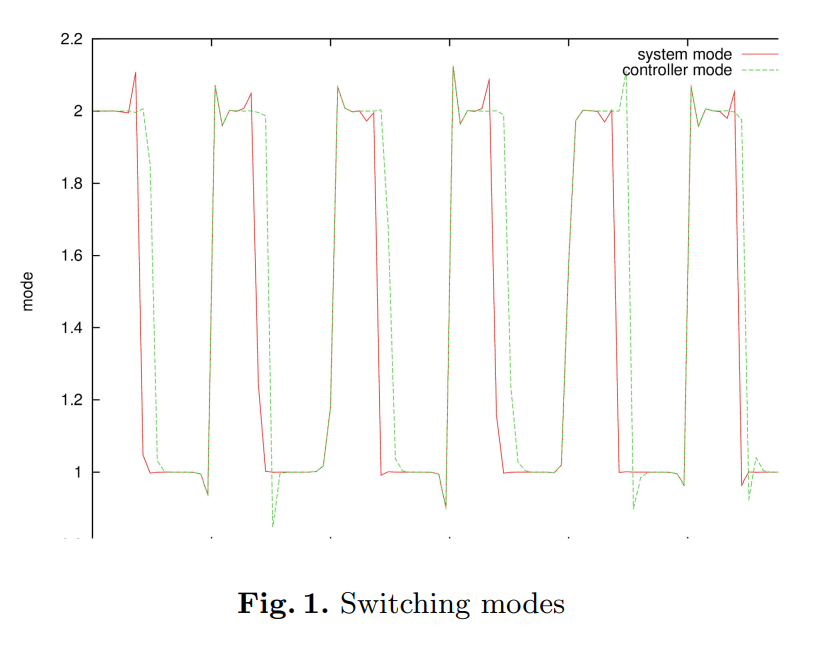
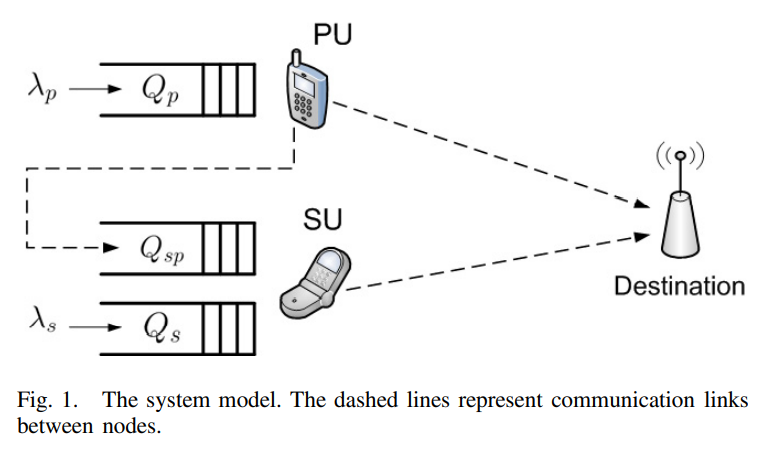
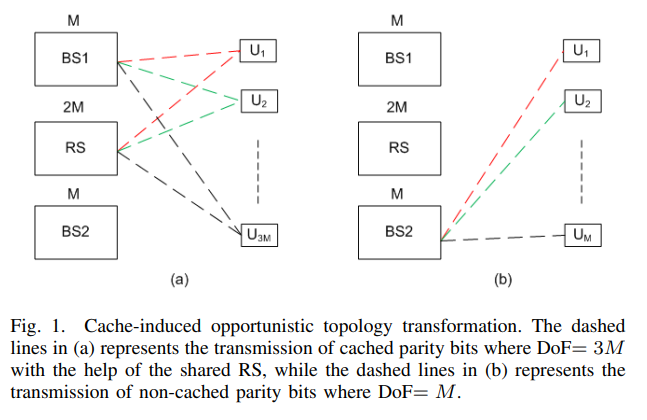
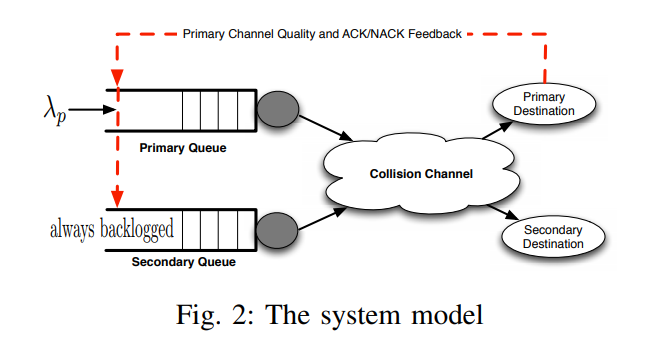
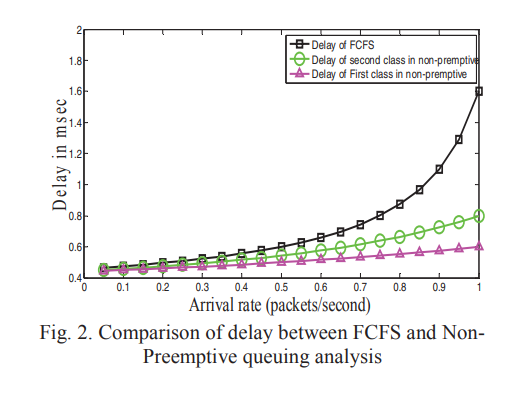
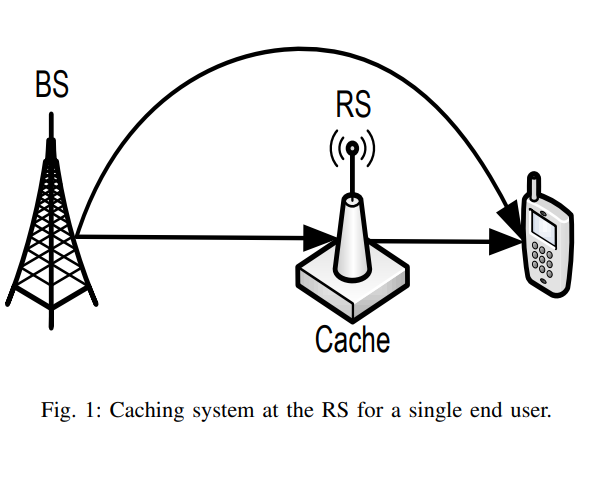
Machine-to-Machine (M2M) has become a generally used term owing to the concept of the Internet of Things (IOT). M2M communications have numerous areas of implementation such as medicine, transportation, environmental monitoring, and smart grids. As the field of its implementation extends, the number of M2M equipment are projected to grow proportionally in the upcoming few years. Current cellular