Predicting non-synonymous single nucleotide variants pathogenic effects in human diseases
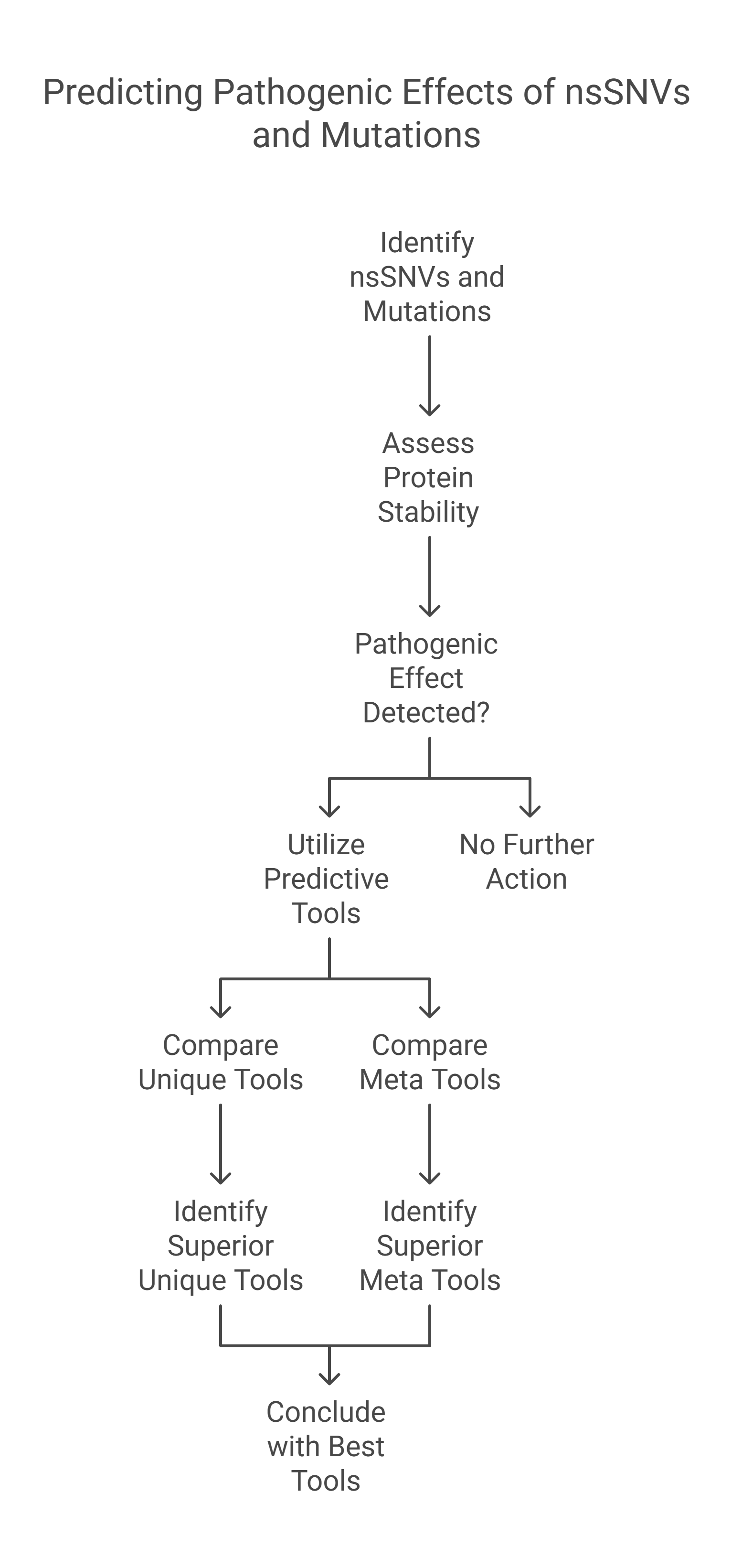
Non-synonymous Single - Nucleotide Variants (nsSNVs) and mutations can create a diversity of the contrary influence of proteins like varying genotype and phenotype of any protein which affects its stability. The alterations in the protein stability may cause diseases. Detecting of nsSNVs and mutations can be a helpful tool in diagnosing diseases at an early stage. The study of singular and consensus tools for predicting pathogenic effects is very essential. Many studies utilized various predicting servers based on distinct Machine Learning Techniques (MLTs). In this research, we conduct a comprehensive study to most of the provided tools or servers that predict disease-causing mutations and the pathogenic effects of nsSNVs with a concentration on the most common. We collected the best unique tools that predict nsSNVs and Meta tools that merge between some of them to enhance the accuracy of the predicting. By comparing the results, we observed that REVEL, CADD, COVEC, Meta-SNP, PAPI, and Condel are better tools than other ensemble tools and PON-P2, SNP dryad, CHASM, PROVEAN, PhD-SNPg, and Mutation Assessor are better tools than other unique tools. The Meta (ensemble) classifiers are better than unique ones as they have the highest overall performance. © 2019 Elsevier Inc. All rights reserved.
Related Publications

Hands-on analysis of using large language models for the auto evaluation of programming assignments