Efficient distributed computation of maximal exact matches
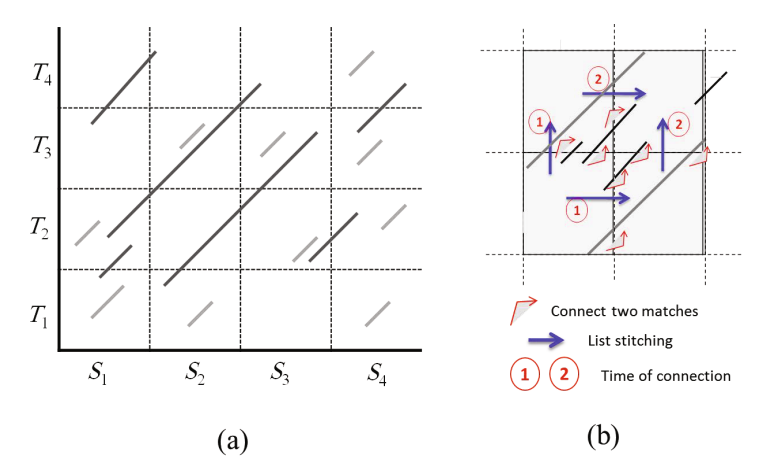
Given two long strings S and T, representing two genomic sequences, and given a user defined threshold ℓ, the problem of computing maximal exact matches (MEMs) is to find each triple (p 1,p 2,l) specifying two matching substrings S[p 1..p 1 + l - 1] = T[p 2..p 2 + l - 1], such that l ≥ ℓ and S[p 1 - 1] ≠ T[p 2 - 1] and S[p 1 + l] ≠ T[p 2 + l]. Computing MEMs is a major problem in bioinformitcs, because it is a primary step in identifying regions of common similarity among genomic sequences. Faster solutions to this problem are still demanded to overcome the ever increasing amount of genomic sequences to be compared to each other. In this paper, we present a parallel version of the MEM algorithm running on a computer cluster. Our experimental results show that our algorithm is efficient and scalable. © 2012 Springer-Verlag.
Related Publications

Hands-on analysis of using large language models for the auto evaluation of programming assignments