


Impact of alignment algorithms on 16S metagenomics analysis
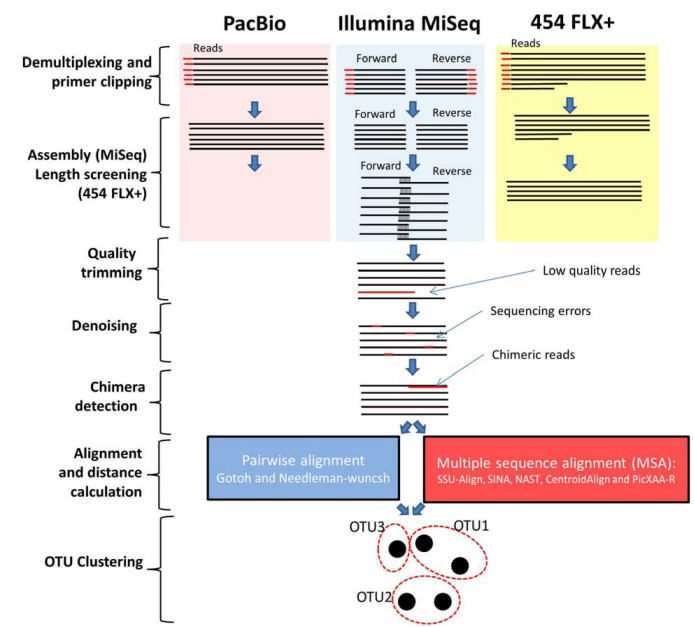
The development of high-throughput sequencing technologies has provided microbial ecologists with an efficient approach to assess bacterial diversity at an unseen depth. In the last year, various platforms have been used for such analysis particularly the Illumina MiSeq, 454 pyrosequencing GS FLX+ and PacBio sequencing platforms. However, analysing such high-throughput data is posing important computational challenges, requiring specialized bioinformatics solutions ending with clustering those sequences into Operational Taxonomic Units (OTUs). Individual algorithms grappling with each of those challenges and numerous efforts have been put to compare them. Nonetheless, there is a need to elucidate the effect of the alignment strategy and subsequently the distance calculation on the OTU-clustering. In this work, a comparative analysis between various alignment algorithms in respect to the produced OTUs is performed. These results suggest that multiple sequence alignment driven distances, particularly those incorporating the secondary structure, are more accurate than pairwise alignment approaches. Additionally, disregarding the computation burden, the de novo multiple sequence aligners are superior to reference based aligners. © 2016 IEEE.