From Reads to Microbial Analysis: An Ensemble Processing Pipeline for 16S Amplicon Sequencing Data (OCToPUS v2)
Objective/Contributions:
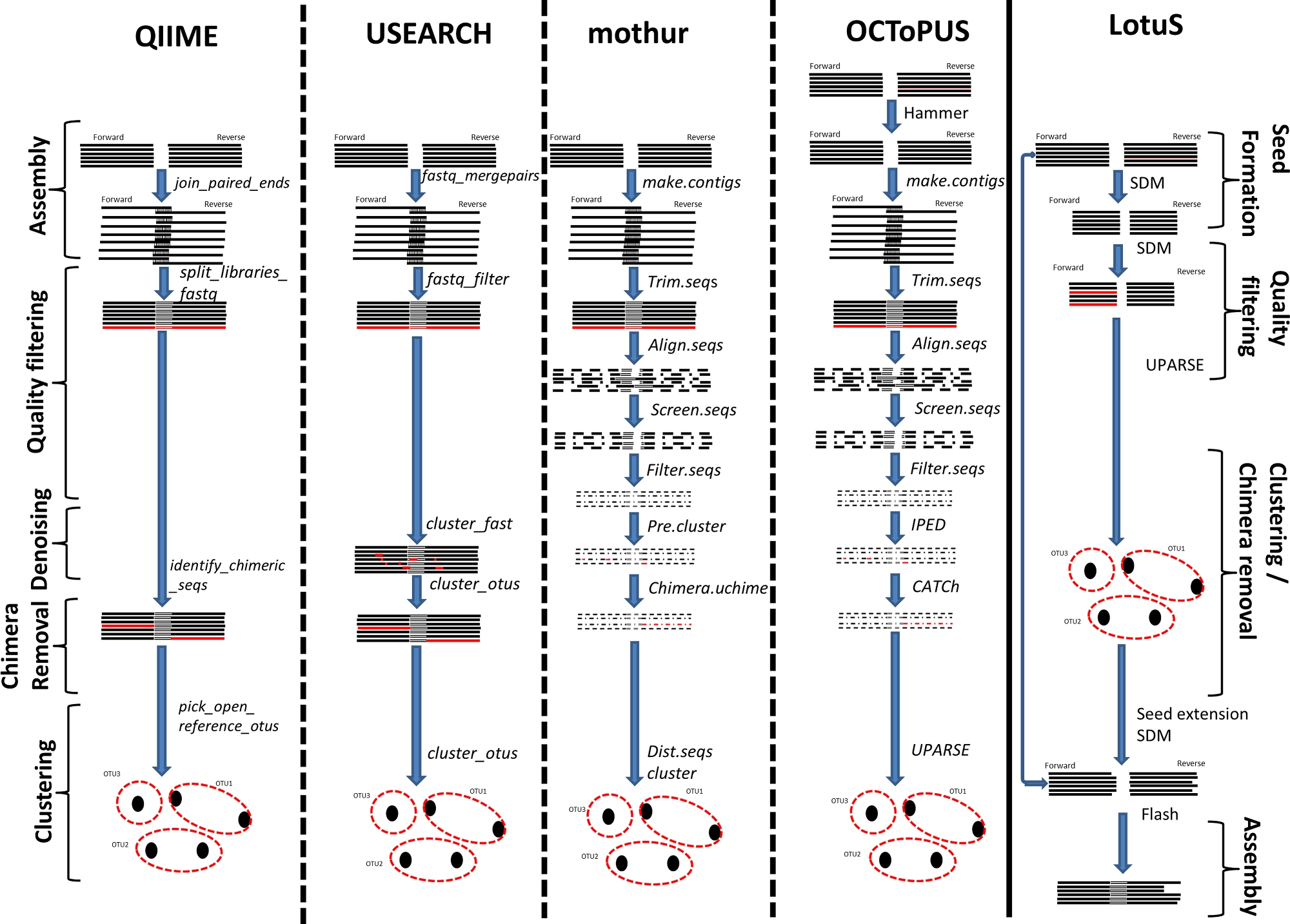
16S microbial profiling came with its own challenges, particularly on the data analysis and interpretation part. This requires specialized bioinformatics solutions at different stages during the processing pipeline, such as assembly of paired-end reads, chimera removal, correction of sequencing errors, and clustering of those sequences into Operational Taxonomic Units (OTUs). Several algorithms were developed to address each one of those challenges, for instance, CATCh (v.1. Mysara et al., 2015) for chimera detection, IPED/NoDe for sequencing error (v.1. Mysara et al., 2016; Mysara, Leys, et al., 2015), DynamiC for OTU-binning (v.1. Mysara, Vandamme, et al., 2017). We have assembled those tools into a single pipeline, named OCToPUS (v.1., Mysara et al., 2017). Although those tools outperformed their alternative at the time of publishing, other more recent tools were developed to better address the advancement in the field. Moreover, the analysis continues beyond the OTU-binning to also cover sub-OTU assignment (what is called Amplicon sequence variants, or ASV), alpha diversity, beta-diversity and hypothesis testing, which takes the raw reads and produces the final report with microbial profiling. So our aim was an enhancement of the already existing unified pipeline “OCToPUS”.
Outcome: Publications
- Goussarov, G., Claesen, J., Mysara, M., Cleenwerck, I., Leys, N., Vandamme, P., & Van Houdt, R. (2022). Accurate prediction of metagenome-assembled genome completeness by MAGISTA, a random forest model built on alignment-free intra-bin statistics. Environmental Microbiomes. https://doi.org/10.1186/s40793-022-00403-7
- Mysara, M., Leys, N., Raes, J., & Monsieurs, P. (2015). NoDe: a fast error-correction algorithm for pyrosequencing amplicon reads. BMC Bioinformatics, 16(1), 88. https://doi.org/10.1186/s12859-015-0520-5
- Mysara, M., Leys, N., Raes, J., & Monsieurs, P. (2016). IPED: a highly efficient denoising tool for Illumina MiSeq Paired-end 16S rRNA gene amplicon sequencing data. BMC Bioinformatics, 17(1), 192. https://doi.org/10.1186/s12859-016-1061-2
- Mysara, M., Njima, M., Leys, N., Raes, J., & Monsieurs, P. (2017). From reads to operational taxonomic units: an ensemble processing pipeline for MiSeq amplicon sequencing data. GigaScience. https://doi.org/10.5524/100265
- Mysara, M., Saeys, Y., Leys, N., Raes, J., & Monsieurs, P. (2015). CATCh, an ensemble classifier for chimera detection in 16S rRNA sequencing studies. Applied and Environmental Microbiology, 81(5), 1573–1584. https://doi.org/10.1128/AEM.02896-14
- Mysara, M., Vandamme, P., Props, R., Kerckhof, F.-M., Leys, N., Boon, N., Raes, J., & Monsieurs, P. (2017). Reconciliation between Operational Taxonomic Units and Species Boundaries. FEMS Microbiology Ecology. https://doi.org/10.1093/femsec/fix029